
Background and Symptoms
Huntington’s disorder (HD) is an inherited condition of the brain that affects 2.71 individuals per 100,000 worldwide, varying by region and an individual’s lineage, making it a very rare disorder (Pringsheim et al., 2012). It stops parts of the brain from working normally over time due to damage to and death of neurones, making it a neurodegenerative disorder. This results in psychological symptoms such as cognitive decline (memory and learning difficulties), behavioural impairment (e.g., mood disorders), and physical symptoms of motor impairment such as chorea (uncontrollable jerky movements), dysphagia (swallowing difficulties), speaking impairment, sluggish movements, and breathing problems. These symptoms gradually worsen over an individual’s lifetime until they’re fatal, with median survival being 24 years from diagnosis and 35 years from symptom onset, and mean age at death being 58 years (Rodrigues et al., 2017).
HD, much like Alzheimer’s, is a disorder that causes dementia, which is characterised by an impaired cognitive ability to concentrate, remember, and make everyday decisions (CDC, 2019). HD and Alzheimer’s also have similar mechanisms of disease, meaning that a better understanding of HD’s pathology could potentially lead to advancements in the research of Alzheimer's and other dementia-causing disorders with similar mechanisms. Alzheimer’s affects over 850,000 people in the UK alone and is predicted to double by 2040. Unlike HD, 99% of cases are caused by aging instead of inherited genetic risk factors (Alzheimer’s Society, 2020; Alzheimer’s Society, No Date; Tanzi, 2012). Therefore, any advancements in the study of HD that lead to advancements in Alzheimer’s research will impact a significantly larger population than only those with HD.
HD is autosomal-dominant, meaning it is caused by a mutation in a non-sex chromosome gene that has a 50% chance of being passed on if one parent has the mutated gene. The mutation is in the HTT gene that codes for the protein huntingtin (Figure 1). Specifically, it occurs in the CAG repeats of the gene, which consists of a cytosine-adenine-guanine repetitive DNA sequence that codes for a polyglutamine amino acid sequence, with glutamine being the corresponding amino acid for these triplet bases. This mutation causes an expansion of these repeats, leading to the production of abnormal, misfolded huntingtin protein (Nopoulos, 2016) (Figure 2).
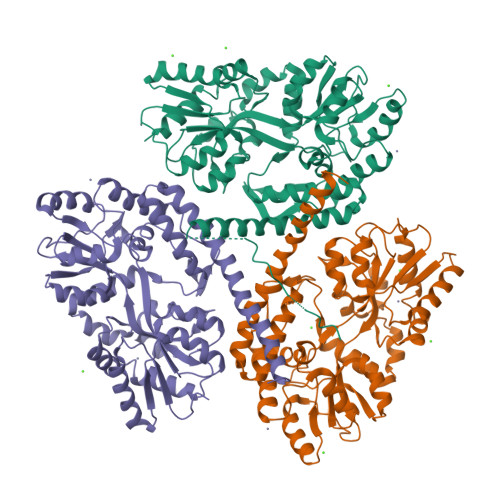
Figure 1. Structure of Native Huntingtin Protein Produced by X-Ray Diffraction. The protein used was produced from the genome of Homo sapiens. The three domains of the cyclic homotrimer protein are coloured green, orange, and purple respectively (Kim et al., 2009).
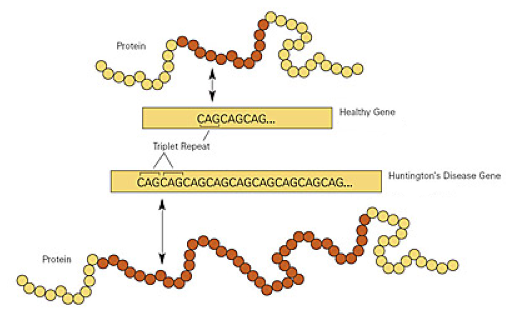
Figure 2. Diagram Showing the Differences in the CAG Repeat Region Between a Healthy and Disease-Causing HTT Gene. The yellow boxes show parts of the CAG repeat region and its expansion in the disease-causing HTT gene (labelled “Huntington’s Disease Gene”). Normal and misfolded / disease-causing proteins are portrayed by yellow (non-repeated) and red (repeated) circles which represent individual glutamine molecules. Arrows show how each “CAG” codes for a single glutamine molecule (NIST, 2011).
The accumulation of these mutant huntingtin (mHTT) proteins leads to a breakdown in regular motor function, cognition, and behaviour (Pagan, Torres-Yaghi and Altshuler, 2017). The mHTT proteins were found to play a key role in mitochondrial dysfunction and the impairment of the electron transport chain, which is a key component of metabolism and energy production in cells. This leads to higher oxidative stress and the release of reactive oxygen species, such as hydrogen peroxide, which ultimately damages cellular machinery and destroys neurones (Liu et al., 2017). It was also found that larger mHTT proteins – with significantly longer polyglutamine sequences – may undergo cleavage and produce shorter protein fragments due to structural breakdown. This leads to the aggregation of these protein fragments to form inclusion bodies in the nuclei (Figure 3), which will disrupt neural activity once reaching neurotoxic levels, eventually leading to cell death (Rubinsztein and Carmichael, 2003).
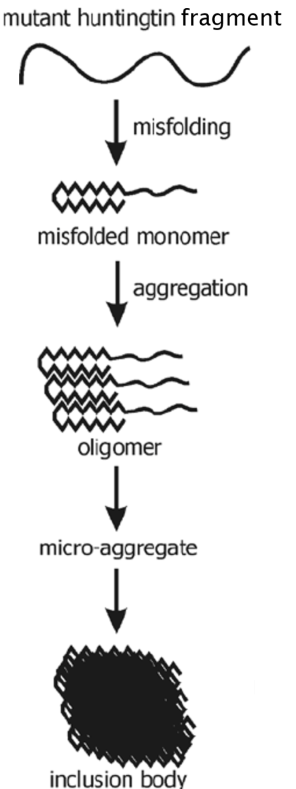
Figure 3. Diagram Showing the Misfolding and Subsequent Aggregation of Mutant Huntingtin Fragments to Form an Inclusion Body (de Pril, 2011).
The disruption of neural activity, and later cell death, leads to the expression of the hallmark symptoms of HD. Late-stage HD can require full-time nursing care because of the severity of these symptoms, which onset most commonly around the age of 40 – though they can rarely start even earlier or later (NHS, 2022).
Prevalence and Inheritance
HD’s prevalence can be higher or lower for a given population depending on the lineage of the population in question. For example, in Europe, North America, and Australia, the overall prevalence is 5.70 per 100,000, with Europe alone having ~10 per 100,000. In Asia, the overall prevalence is 0.40 per 100,000 (Pringsheim et al., 2012). Not only does this portray the rarity of HD, but it also shows how genetic disorders can have significantly different prevalence rates around the world because of a region’s population’s lineage.
Although there has been an increase in prevalence rates worldwide, for example, a 15-20% increase per decade in studies in Australia, North America, and Western Europe (Rawlins et al., 2016), this increase cannot be attributed solely to any increase in incidence. Factors such as better diagnostic screening, public willingness to get tested, and more funding play major roles in the reported increase in prevalence (Wexler et al., 2016). Another factor to note for recent prevalence and incidence rates is that HD does not receive equal attention across the world. Some countries, such as those in the UK, for example, have the funding and research power needed to produce modern diagnostic and treatment techniques. Some countries, such as those attributed to being third-world countries, may not have the same funding and research power, which means large numbers of patients are missed for diagnosis and treatment.
In terms of incidence rates, in Europe, North America, and Australia, incidence rates ranged from 0.11 to 0.8 per 100,000 per year. In Asia, incidence rates ranged from 0.046 to 0.16 per 100,000 per year. According to one study’s examination of these incidence rates, no trends towards a decrease or increase in incidence over time were observed, meaning incidence has largely remained the same but prevalence has increased (Pringsheim et al., 2012). Interestingly, another study (Roos, 2010) only examined incidence in 45- to 64-year-olds and revealed an incidence rate of 0.8 per 100,000 per year (Mercy et al., 2008). This relatively high incidence rate could be explained by the fact that symptoms often develop between the ages of 30 and 50, so any symptoms that have developed enough to be noticed and attributed to HD may be spotted in the study’s range of 45- to 64-year-olds, leading to a diagnosis and an increased incidence rate for this age range. However, this study was only conducted in Cambridgeshire, United Kingdom, so this incidence rate may not represent 45- to 64-year-olds in Europe, North America, Australia, and Asia as accurately.
As mentioned previously, due to HD being a dominant genetic disorder, if one parent has the disorder, then their children have a 50% chance of having the mHTT gene (Walker, 2007). However, there are certain thresholds associated with the number of CAG repeats that influence the inheritance of HD - if the HTT gene contains ≤26 CAG repeats, it is considered normal, but if it contains ≥40, it is considered to be associated with HD, and the individual will likely have the disorder (Myers, 2004). Furthermore, if the gene contains over 28 CAG repeats, it may not be associated with HD but instead could pose a risk to future offspring. This is because CAG repeats over 28 become increasingly more unstable with length, leading to further expansion. This means that, while both parents may not have an HTT gene of abnormal length if one parent has an HTT gene with over 28 CAG repeats, their children may still have a chance of acquiring an mHTT gene, and by extension, HD (Walker, 2007). This increasing length through generations leads to earlier age onset due to larger quantities of neurotoxic polyglutamine being produced, increasing the disease’s severity. This is known as genetic anticipation (Dayalu and Albin, 2015).
Diagnosis and Treatment
Because HD is monogenetic – meaning it is caused by a single faulty gene – diagnosis is relatively straightforward. The diagnostic test consists of a blood test that, when analysed, allows for the quantity of CAG repeats in each HTT allele to be determined (Myers, 2004). This means the test can determine the probability of developing HD in the future because a result of ≥40 implies a 100% risk, 36-39 implies a 60-70% risk, 27-35 implies a 0% personal risk - with risk to offspring acquiring the mHTT gene - and ≤26 implies a 0% risk (de Die-Smulders et al., 2013). Individuals in the UK who wish to get the test, such as after finding out one of their parents has HD and wish to know their probability of developing HD, can visit a regional genetics clinic once they have arranged the appointment with their GP. Patients who are awaiting testing at a clinic usually have a few counselling sessions to discuss testing implications and any concerns before deciding to follow through on testing. Follow-up counselling and support are also offered once a patient receives their test results (Huntington’s Disease Association, No Date). Prenatal testing also exists to predict the foetus’s risk of developing HD, however, this can lead to an ethical dilemma around the act of testing and knowing, as well as the possibility of selective abortion (Spurdle et al., 1991). Other potential options for individuals with HD who want children include pre-implantation genetic diagnosis, which is a form of IVF where the zygote is genetically tested for HD prior to implantation, egg or sperm donations, adoption, or conceiving naturally without any testing, although this would require accepting the risk of the child inheriting HD (Huntington’s Disease Association, No Date).
When a diagnosis is given to a patient, potential treatments will be discussed. These treatments are not aimed at curing, as a cure has not yet been discovered, but are instead aimed at regulating symptoms. These consist of nutrition management to counter weight loss due to dysphagia, thickening agents for liquids to make them easier and safer to swallow, and reminders from carers to eat slowly to prevent choking (Walker, 2007). As for medications, tetrabenazine is used to treat chorea. It does this by lowering dopamine levels, which reduces nerve cell communication and ultimately chorea. Consequently, the lowered dopamine levels are reflected in an increased risk of suicidal ideations, although tetrabenazine is not the only factor behind this increased risk (FDA, 2008).
Current Research
There are a wide variety of research studies taking place to combat HD. In 2020 alone, there were 200 clinical trials based on potential therapies and biomarkers (Clinicaltrials.gov, 2020). Some research studies are actively looking into reducing the levels of mHTT protein by looking for ways to increase the rate at which cells dispose of it. These studies have shown that mHTT levels can be reduced through the application of autophagy-inducing factors, both natural and artificial (Djajadikerta et al., 2020). Autophagy is the cells’ processes of recycling old and damaged cellular parts, such as mHTT and its fragments (Klionsky, 2008). Other studies have demonstrated that gene silencing is a potential route as it takes advantage of the fact that HD is monogenic. They aim to reduce the production of mHTT protein by application of ASOs – antisense oligosaccharides – which bind to mHTT mRNA and prevent its transcription, effectively reducing the protein concentrations and halting its neurodegenerative effects. (Munoz-Sanjuan and Bates, 2011). Another study investigated the potential application of stem-cell therapy for the regeneration of damaged neurons by transplanting stem cells into afflicted regions of the brain (Clelland, Barker and Watts, 2008).
A novel staging system for HD was also created in a similar way to how cancer is staged from 0 up to 4. Huntington’s Disease Integrated Staging System (HD-ISS) was made with the intention of assisting clinical trials target the earliest stages of HD, and is based on biological, functional, and clinical characteristics of HD. It is also the first staging system ever created for a genetic neurological disorder (Tabrizi et al., 2022).
A recent study discovered that autophagy is impaired in people with HD, leading to a build-up of biochemical waste in affected neurones, eventually causing cell death. Using a technique developed by the researchers, they were able to reprogram adult skin cells from someone symptomatic with HD into medium spiny neurones using specific signalling molecules. It was necessary to use adult skin cells as stem cells would only be useful for studying a cell’s early developmental state, whereas HD is most commonly symptomatic in adulthood. The HD-afflicted medium spiny neurones were observed producing chemicals that set off a cascade of events, including autophagy impairment. The researchers also identified a chemical, ‘G2’, using high throughput screening of a database of autophagy enhancers, added this to the neurones, and observed how G2 enhanced autophagy and prevented cell death (Oh et al., 2022).
A research study investigating the drug pridopidine as an HD treatment has also shown promise. The drug helps to encourage the production of a neuroprotective protein known as a brain-derived neurotrophic factor, which is found at reduced levels in people with HD. The protein has been described as a preventative in mHTT-induced cell death and also leads to a reduction in the HD-impairment of cellular processes. A PET imaging study was used to study the drug’s occupation of the brain receptor ‘S1R’ and showed that it occupied 90% of them. These results were used to support the testing of the drug’s appropriate dose in the trial (Huntington Study Group, 2022; Reilmann et al., 2021).
Conclusion
In conclusion, Huntington’s disorder is caused by a mutation in the HTT gene leading to defective huntingtin protein that produces neurotoxic fragments of polyglutamine. This neurotoxicity leads to the expression of nerve-related psychological and physical symptoms due to neural damage in the brain. It is a relatively rare disorder with 10 in 100,000 Europeans being afflicted, despite being autosomal-dominant. It is diagnosed through a blood test that determines CAG repeat quantities in alleles to determine susceptibility. It is treated through a mix of therapies, dietary and habit changes, and medication. Current research aims to combat this disorder through gene silencing, neural regeneration, and autophagic-induction. Research within the last year has led to the creation of a novel staging system to help with trialling more effective treatments, the discovery of Huntington’s disorder impairing autophagy processes, and the clinical trialling of a new and promising drug.
References
Alzheimer’s Society. (2020). ‘Alzheimer’s Society’s view on demography’. Available at: https://www.alzheimers.org.uk/about-us/policy-and-influencing/what-we-think/demography#:~:text=Research%20conducted%20shows%20that%2C%20in,the%20current%20rate%20of%20prevalence (Accessed 04 June 2023).
Alzheimer’s Society. (No Date). ‘Is dementia hereditary?’ Available at: https://www.alzheimers.org.uk/about-dementia/risk-factors-and-prevention/is-dementia-hereditary (Accessed 04 June 2023).
Balogun, B., Sutherland, N. and Powell, T. (2022). ‘Huntington’s disease’. Number CDP 2022/0191. Available at: https://researchbriefings.files.parliament.uk/documents/CDP-2022-0191/CDP-2022-0191.pdf (Accessed 26 May 2023).
CDC. (2019). ‘About Dementia’. Alzheimer’s Disease and Related Dementias . Available at: https://www.cdc.gov/aging/dementia/index.html#:~:text=Dementia%20is%20not%20a%20specific,a%20part%20of%20normal%20aging (Accessed 04 June 2023).
Clelland, C.D., Barker, R.A. and Watts, C. (2008). ‘Cell therapy in Huntington disease’. Neurosurgical Focus , 24(3-4), p.E9.
Clinicaltrials.gov. (2020). ‘Search of: Huntington Disease - List Results - ClinicalTrials.gov’. Available at: https://clinicaltrials.gov/ct2/results?cond=Huntington+Disease&term=&cntry=&state=&city=&dist= (Accessed 10 February 2022).
Dayalu, P. and Albin, R.L. (2015). ‘Huntington Disease’. Neurologic Clinics , 33(1), pp.101–114.
de Die-Smulders, C.E.M., de Wert, G.M.W.R., Liebaers, I., Tibben, A. and Evers-Kiebooms, G. (2013). ‘Reproductive options for prospective parents in families with Huntington’s disease: clinical, psychological and ethical reflections’. Human Reproduction Update , 19(3), pp.304–315.
Djajadikerta, A., Keshri, S., Pavel, M., Prestil, R., Ryan, L. and Rubinsztein, D.C. (2020). ‘Autophagy Induction as a Therapeutic Strategy for Neurodegenerative Diseases’. Journal of Molecular Biology , 432(8), pp.2799–2821.
FDA (2008). ‘FDA Approves First Drug for Treatment of Chorea in Huntington’s Disease’. Available at: https://web.archive.org/web/20080821020643/https:/www.fda.gov/bbs/topics/NEWS/2008/NEW01874.html (Accessed 10 February 2022).
Frank, S. (2013). ‘Treatment of Huntington’s Disease’. Neurotherapeutics , 11(1), pp.153–160.
Huntington Study Group (2022). ‘PRidopidine Outcome On Function in Huntington Disease (PROOF-HD)’. Available at: https://huntingtonstudygroup.org/proof-hd/ (Accessed 05 June 2023).
Huntington’s Disease Association. (No Date). ‘Genetic testing’. Available at: https://www.hda.org.uk/information-and-support/getting-help/genetic-testing/ (Accessed 05 June 2023).
Huntington’s Disease Association. (No Date). ‘Starting a family’. Available at: https://www.hda.org.uk/information-and-support/getting-help/starting-a-family/ (Accessed 05 June 2023).
Kim, M.W., Chelliah, Y., Kim, S.W., Otwinowski, Z. and Bezprozvanny, I. (2009). ‘Secondary Structure of Huntingtin Amino-Terminal Region’. Structure , 17(9), pp.1205–1212.
Klionsky, D.J. (2008). ‘Autophagy revisited: A conversation with Christian de Duve’. Autophagy , vol 4, pp.740-743.
Liu, Z., Zhou, T., Ziegler, A.C., Dimitrion, P. and Zuo, L. (2017). ‘Oxidative Stress in Neurodegenerative Diseases: From Molecular Mechanisms to Clinical Applications’. Oxidative Medicine and Cellular Longevity , pp.1–11.
Mercy, L., Hodges, J.R., Dawson, K.S., Barker, R.A. and Brayne, C. (2008). ‘Incidence of early-onset dementias in Cambridgeshire, United Kingdom’. Neurology , 71(19), pp.1496–1499.
Munoz-Sanjuan, I. and Bates, G.P. (2011). ‘The importance of integrating basic and clinical research toward the development of new therapies for Huntington disease’. Journal of Clinical Investigation , 121(2), pp.476–483.
Myers, R.H. (2004). ‘Huntington’s disease genetics’. NeuroRX , 1(2), pp.255–262.
NHS (2022). ‘Symptoms - Huntington’s disease’. Available at: https://www.nhs.uk/conditions/huntingtons-disease/ (Accessed 10 February 2022).
NIHR (2022). ‘NIHR Annual Report 2020/2021’. Available at: https://www.nihr.ac.uk/documents/about-us/our-contribution-to-research/research-performance/Final-NIHR-Annual-report%2020_21.pdf (Accessed 26 May 2023).
NIST. (2011). ‘New NIST SRM Helps Improve Diagnosis of Huntington’s Disease’. Available at: https://www.nist.gov/news-events/news/2011/04/new-nist-srm-helps-improve-diagnosis-huntingtons-disease (Accessed 27 May 2023).
Nopoulos, P. (2016). ‘Huntington disease: a single-gene degenerative disorder of the striatum’. Dialogues in Clinical Neuroscience , 18(1), pp.91–98.
Oh, Y.M., Lee, S.W., Kim, W.K., Chen, S., Church, V.A., Cates, K., Li, T., Zhang, B., Dolle, R.E., Dahiya, S., Pak, S.C., Silverman, G.A., Perlmutter, D.H. and Yoo, A.S. (2022). ‘Age-related Huntington’s disease progression modeled in directly reprogrammed patient-derived striatal neurons highlights impaired autophagy’. nature neuroscience , 25(11), pp.1420–1433.
Pagan, F., Torres-Yaghi, Y. and Altshuler, M. (2017). ‘The diagnosis and natural history of Huntington disease’. Handbook of clinical neurology , pp.63–67.
Pril, R. (2011). ‘The ubiquitin proteasome system in Huntington disease: impairment of the proteolytic machinery aggravates huntingtin aggregation and toxicity’. Protection.
Pringsheim, T., Wiltshire, K., Day, L., Dykeman, J., Steeves, T. and Jette, N. (2012). ‘The incidence and prevalence of Huntington’s disease: A systematic review and meta-analysis’. Mov Disord , 27(9), pp.1083–1091.
Rawlins, M.D., Wexler, N.S., Wexler, A.R., Tabrizi, S.J., Douglas, I., Evans, S.J.W. and Smeeth, L. (2016). ‘The Prevalence of Huntington’s Disease’. Neuroepidemiology , 46(2), pp.144–153.
Reilmann, R., Rosser, A., Kostyk, S., Geva, M., McGarry, A., Cohen, Y., Gershoni-Emek, N., Mehra, M., Olanow, C.W., Kieburtz, K., Hayden, M.R. and Feigin, A. (2021). ‘F41 The proof-hd phase 3 study: pridopidine’s outcome on function in huntington disease (PROOF)’. Neurology, Neurosurgery & Psychiatry , 92(1).
Rodrigues, F.B., Abreu, D., Damásio, J., Gonçalves, N., Correia-Guedes, L., Coelho, M., Ferreira, J.J. and REGISTRY Investigators of the European Huntington's Disease Network (2017). ‘Survival, Mortality, Causes and Places of Death in a European Huntington’s Disease Prospective Cohort’. Movement Disorders Clinical Practice , 4(5), pp.737–742.
Roos, R.A.C. (2010). ‘Huntington’s disease: a clinical review’. Orphanet J Rare Dis , 5(1), pp.40.
Rubinsztein, D.C. and Carmichael, J. (2003). ‘Huntington’s disease: molecular basis of neurodegeneration’. Expert Reviews in Molecular Medicine , 5(20), pp.1–21.
Spurdle, A., Kromberg, J., Rosendorff, J. and Jenkins, T. (1991). ‘Prenatal diagnosis for huntington’s disease: A molecular and psychological study’. Prenat Diagn , 11(3), pp.177–185.
Tabrizi, S.J., Schobel, S., Gantman, E.C., Mansbach, A., Borowsky, B., Konstantinova, P., Mestre, T.A., Panagoulias, J., Ross, C.A., Zauderer, M., Mullin, A.P., Romero, K., Sivakumaran, S., Turner, E.C., Long, J.D., Sampaio, C. and the Huntington's Disease Regulatory Science Consortium (2022). ‘A biological classification of Huntington’s disease: the Integrated Staging System’. Neurology , 21(7), pp.632–644.
Tanzi, R.E. (2012). ‘The Genetics of Alzheimer Disease’. Cold Spring Harbor Perspectives in Medicine , 2(10), pp.a006296–a006296.
Walker, F.O. (2007). ‘Huntington’s disease’. The Lancet , 369(9557), pp.218–228.
Wexler, N.S., Collett, L., Wexler, A.R., Rawlins, M.D., Tabrizi, S.J., Douglas, I., Smeeth, L., and Evans, S.J. (2016). ‘Incidence of adult Huntington’s disease in the UK: a UK-based primary care study and a systematic review’. BMJ Open , 6(2), pp.e009070–e009070.
© Luke David Wheeler. This article is licensed under a Creative Commons Attribution 4.0 International Licence (CC BY).